

A decade ago, genome sequencing revealed a big surprise: about 50 percent of human cancers are linked to mutations in what are known as epigenetic regulators, which control the activity of genes.
In a new study in Cell Chemical Biology, a team of scientists led by Oliver Bell from USC and Stephen V. Frye from the University of North Carolina at Chapel Hill developed a new drug-like molecule that can counteract the effects of mutated epigenetic regulators, which are known to drive certain types of cancer including lymphoma.
In healthy cells, epigenetic regulators play an essential role: turning on and off the activity of hundreds of genes in the precisely orchestrated sequence that directs normal human development.
One of these epigenetic regulators, EZH2, controls the transient inactivation of specific genes in order to permit the maturation of immune cells. However, mutated EZH2 may cause persistent repression of these genes, thus preventing the immune cells from developing normally and ultimately leading them to transform into cancerous malignancies.
The good news is that in contrast to many other types of mutations, cancer-causing mutations in epigenetic regulators are potentially reversible by therapeutic drugs. With this in mind, first author Junghyun L. Suh and the research team set out to design a drug-like molecule to reverse the cancer-causing gene repression by EZH2.
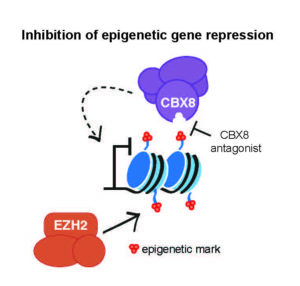 Suh and her colleagues started by considering the mechanism by which EZH2 controls gene repression. EZH2 acts as a “writer” that marks which genes will be repressed. A second epigenetic regulator called CBX8 serves as a “reader” that interprets these repressive marks, and recruits additional regulatory machinery that actually turns off the genes.
Suh and her colleagues started by considering the mechanism by which EZH2 controls gene repression. EZH2 acts as a “writer” that marks which genes will be repressed. A second epigenetic regulator called CBX8 serves as a “reader” that interprets these repressive marks, and recruits additional regulatory machinery that actually turns off the genes.
Compared to the writer, the reader CBX8 seems to be equally critical for the proliferation of cancer cells, but is more dispensable for the function of healthy cells. This means that drugs targeting the reader would be expected to have fewer toxic side effects on the healthy cells throughout a patient’s body.
To specifically target CBX8, the researchers first engineered mouse stem cells in which they could easily screen a large number of drug-like molecules. These engineered stem cells relied on CBX8 reading the marks deposited by EZH2 to repress a gene producing a visible green fluorescent protein (GFP). If the stem cells showed activation of the telltale green glow, the scientists knew that a drug-like molecule had successfully prevented CBX8 from reading the repressive marks.
The researchers then parlayed their knowledge of CBX8 into several iterations of drug-like molecules that targeted this particular reader. They took into account CBX’s intricate protein structure, as well as the way that it binds to DNA and reads repressive marks. When they had succeeded in synthesizing a potent molecule that worked well in the engineered mouse cells, they moved on to testing in human cancer cells.
“When we exposed human lymphoma and colorectal cancer cells to our newly synthesized drug-like molecule in the laboratory, the malignant cells ceased to proliferate and began to behave more like healthy cells,” said the study’s co-corresponding author Oliver Bell, who is an assistant professor of biochemistry and molecular medicine, and stem cell biology and regenerative medicine at the Keck School of Medicine of USC, and a member of the USC Norris Comprehensive Cancer Center.
“Our CBX8-targeted molecule has the most powerful effect that we’ve seen so far in terms of blocking the reader’s function,” added co-corresponding author Stephen V. Frye, who is the Fred Eshelman Distinguished Professor and Co-director of the Center for Integrative Chemical Biology and Drug Discovery at the University of North Carolina at Chapel Hill. “This opens a path to exploring related cancer therapies, as well as to enhancing our understanding of epigenetic regulation in normal human development.”
Additional co-authors include: Daniel Bsteh, Yibo Si, Roy Lau, Shivani Soni, Shannon M. Mumenthaler, and Heinz-Josef Lenz from USC; Bryce Hart, Justin M. Rectenwald, Jacqueline L. Norris, Stephanie H. Cholensky, Jessica D. Umana, Dongxu Li, Brian Hardy, Gang Greg Wang, Ken H. Pearce, Lindsey I. James, and Dmitri B. Kireev from the University of North Carolina at Chapel Hill; Tyler M. Weaver from the University of Iowa; Carina Pribitzer from the Institute of Molecular Biotechnology of the Austrian Academy of the Sciences at the Vienna Biocenter in Austria; Heather Ogana and Yong-mi Kim from Children’s Hospital Los Angeles and USC; Cari Sagum and Mark T. Bedford at the University of Texas MD Anderson Cancer Center; and Catherine A. Musselman from the University of Iowa and the University of Colorado Anschutz Medical Campus.
Eighty percent of this work was supported by U.S. federal funding from the National Institutes of Health (NIH), including the National Cancer Institute (R01CA218392, P30CA014089, P30CA086862, P30CA016086), the National Institute of General Medical Sciences (R01GM100919, R35GM128705, R01GM132299), and the National Institute on Drug Abuse (R61DA047023). The remaining 20 percent of the work was funded by the New Frontiers Group of the Austrian Academy of Sciences (NFG-05), the USC Norris Comprehensive Cancer Center, the Gloria Borges WunderGlo Foundation, the Gene Gregg Pancreas Cancer Research Fund, The Holden Comprehensive Cancer Center at The University of Iowa, the UNC Lineberger Comprehensive Cancer Center, the UNC Eshelman Institute for Innovation (RX0351210, RX03712105), and the Cancer Prevention and Research Institute of Texas (RP180804).